
Bone Marrow Failure Syndrome: A Complete Guide
Know about the bone marrow failure syndrome, what it is, risks, cause, signs, symptoms, diagnosis, treatment options, and living well with the syndrome.

Written by Dr. Dhankecha Mayank Dineshbhai
Reviewed by Dr. Md Yusuf Shareef MBBS, Advanced Certificate Course in Dermatology
Last updated on 13th Jan, 2026

Introduction
Bone marrow failure syndrome describes a group of conditions where the bone marrow cannot make enough healthy blood cells. That shortfall can lead to anaemia (low red cells), infections (low white cells), and bleeding (low platelets). While “bone marrow failure” sounds rare and frightening, many people do well with timely diagnosis and the right treatment plan. Some causes are acquired later in life—like aplastic anaemia and myelodysplastic syndromes while others are inherited, such as Fanconi anaemia or Diamond-Blackfan anaemia. Because symptoms can be subtle at first—fatigue, frequent infections, easy bruising recognising warning signs and getting the correct tests early is essential.
In this guide to bone marrow failure syndrome, you’ll learn how healthy marrow works, what can go wrong, the main types, symptoms to watch, how doctors diagnose the condition, and current treatments ranging from supportive care to immunotherapy and stem cell transplant. We’ll also offer practical tips for daily living, discuss long-term outlook, and highlight promising research. Whether you’re newly diagnosed, supporting a loved one, or just trying to understand your lab results, this clear, patient-centred overview is here to help.
Consult a Top General Practitioner for Personalised Advice
What Is Bone Marrow Failure Syndrome?
Bone marrow is the factory for your blood. Inside your bones, stem cells divide and mature into red cells (carry oxygen), white cells (fight infection), and platelets (control bleeding). Bone marrow failure syndrome occurs when that factory slows or stops, leading to too few cells in one or more of those lines (pancytopenia is when all three are low). This is different from isolated anaemia due to iron or vitamin deficiency, where the marrow often works harder to compensate; in marrow failure, the root problem is the stem cells or their environment.
Two broad categories exist: acquired and inherited.
- Acquired causes develop over time—commonly aplastic anaemia (an immune attack on stem cells) or myelodysplastic syndromes (MDS), in which damaged stem cells produce abnormal, ineffective blood cells and can sometimes progress to leukaemia. Another acquired condition, paroxysmal nocturnal haemoglobinuria (PNH), can overlap with aplastic anaemia and causes red blood cells to break down due to missing protective proteins.
- Inherited bone marrow failure syndromes, such as Fanconi anaemia, Diamond-Blackfan anaemia, Shwachman-Diamond syndrome, and dyskeratosis congenita (a telomere biology disorder), stem from genetic changes that impair DNA repair, ribosome function, or telomere maintenance. Those genetic issues often bring extra-medullary features (skin/hair changes, skeletal differences, pancreatic or lung issues) that can hint at the diagnosis. Understanding whether marrow failure is acquired or inherited shapes both treatment decisions and family counselling.
Types of Bone Marrow Failure
- Aplastic anaemia (AA) is a classic acquired marrow failure: the marrow becomes “empty” (hypocellular) and stops producing enough cells. Severity is graded by the depth of low counts and neutropenia. Incidence in Western countries is roughly 2 per million per year, higher in parts of Asia (4–7 per million). Standard treatments include immunosuppressive therapy (horse anti-thymocyte globulin plus cyclosporine) and hematopoietic stem cell transplantation (HSCT), with donor choice guided by age and severity. Response rates to ATG/cyclosporine are often 60–70%, and adding eltrombopag has improved outcomes in some studies.
- Myelodysplastic syndromes (MDS) are clonal disorders, more common with age (median ~70 years), where the marrow is usually cellular but ineffective and dysplastic. Patients can be asymptomatic or have anaemia, infections, and bleeding. MDS carries a risk of evolving into acute myeloid leukaemia. Doctors use risk systems like IPSS-R to guide treatment: supportive care and growth factors for lower-risk disease; hypomethylating agents (azacitidine/decitabine), lenalidomide for del(5q), and transplant for higher-risk disease or fit candidates.
- PNH arises when stem cells acquire a PIGA gene mutation, preventing cells from expressing protective proteins (CD55/CD59). That leads to complement-mediated hemolysis, anaemia, clotting risk, and sometimes abdominal pain or dark urine. PNH frequently overlaps with aplastic anaemia; flow cytometry confirms the diagnosis. Targeted complement inhibitors are now standard in many settings.
Inherited syndromes include:
- Fanconi anaemia: DNA repair defect; birth differences, short stature, cancer risk (especially leukaemias/head and neck cancers), and progressive marrow failure. HSCT is often needed. [8]
- Diamond-Blackfan anaemia: ribosomal defect; typically presents in infancy with severe anaemia; may require transfusions, steroids, or HSCT.
- Shwachman-Diamond syndrome: marrow failure plus pancreatic insufficiency and skeletal issues.
- Dyskeratosis congenita/telomere biology disorders: very short telomeres; nail/skin changes, pulmonary and liver disease, and marrow failure; special transplant conditioning is often required.
- Presentations differ between children and adults, and genetic testing can be decisive in inherited cases.
Signs, Symptoms, and Complications
Symptoms often reflect which cell line is low:
- Anaemia (low red cells): fatigue, weakness, shortness of breath, pale skin, headaches, chest discomfort with exertion.
- Neutropenia (low neutrophils): frequent or unusual infections, non-healing sores, fevers without an obvious cause. In severe neutropenia, even minor infections can escalate quickly. [2][3]
- Thrombocytopenia (low platelets): easy bruising, petechiae (tiny red spots), nosebleeds, bleeding gums, heavy periods, or prolonged bleeding from cuts.
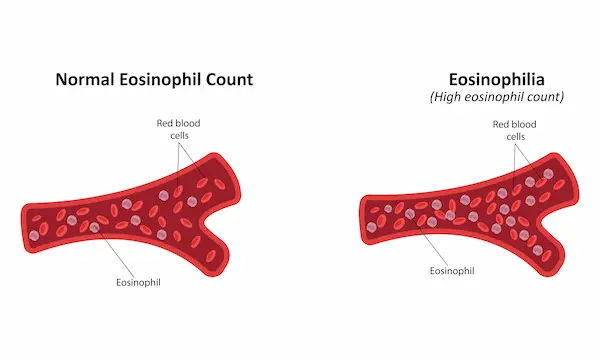
Some conditions carry distinct features: PNH may cause dark morning urine (hemoglobinuria), abdominal pain, or blood clots in unusual veins. Inherited syndromes may include short stature, fingernail changes, skin pigmentation, or skeletal/pancreatic issues. Complications can include severe infections (a leading cause of morbidity with neutropenia), significant bleeding, iron overload from repeated transfusions, and, in some cases, evolution to MDS or leukaemia.
Red flags and when to seek urgent care:
- Persistent fever (above 38°C/100.4°F), especially with known neutropenia
- Uncontrolled bleeding or black/tarry stools
- Sudden chest pain, severe headache, or shortness of breath
- Marked fatigue or fainting spells
If symptoms persist beyond two weeks, consult a doctor online with Apollo 24|7 for further evaluation. If you have a high fever, severe bleeding, or breathing difficulty, go to the emergency department immediately. Early attention can prevent serious complications.
Causes and Risk Factors
- In aplastic anaemia, the most common mechanism is an autoimmune attack on hematopoietic stem cells, often without a single identifiable trigger. Environmental and medication exposures can injure marrow: benzene (industrial solvent), certain chemotherapy agents, radiation, and some antibiotics or antithyroid drugs have been implicated. Viral infections—including hepatitis, EBV, parvovirus B19, and HIV can precede marrow failure; in “hepatitis-associated aplastic anaemia,” severe marrow failure appears weeks to months after acute hepatitis. Rarely, pregnancy is the setting for the onset. Smoking, older age (for MDS), and prior chemo/radiation increase risk.
- Inherited bone marrow failure syndromes arise from germline mutations affecting DNA repair (Fanconi anaemia), ribosome biogenesis (Diamond-Blackfan anaemia), telomere maintenance (dyskeratosis congenita), or other pathways (Shwachman-Diamond syndrome). Family history of early bone marrow failure, unexplained cytopenias, or certain cancers can be a clue. Children can present with congenital anomalies, growth issues, or organ involvement that point toward specific syndromes—data that helps doctors decide which genetic tests to order.
- Understanding cause matters because it changes treatment: autoimmune aplastic anaemia often responds to immunosuppression; MDS is a clonal disorder managed by risk stratification; inherited syndromes usually require specialised transplant conditioning and long-term cancer surveillance. If your condition does not improve after trying initial measures or if your doctor suspects an inherited syndrome, book a physical visit to a doctor with Apollo 24|7 to coordinate genetic testing and comprehensive care.
Diagnosis: Tests You May Need
- The evaluation starts with a complete blood count (CBC) and reticulocyte count to measure how many cells are being produced. Additional labs check iron, ferritin (iron storage), vitamin B12, and folate to rule out nutritionally driven anaemia. Infectious workup (hepatitis viruses, HIV, EBV, parvovirus) may be ordered when history suggests a trigger. Peripheral blood smear often shows clues (dysplasia in MDS, hemolysis in PNH). Apollo 24|7 offers convenient home collection for tests like CBC, iron studies, vitamin B12, and folate—helpful if you’re monitoring trends over time.
- Definitive diagnosis usually requires a bone marrow aspirate and biopsy. In aplastic anaemia, the marrow is hypocellular without malignant cells; in MDS, the marrow is often cellular but shows dysplasia and cytogenetic abnormalities. Cytogenetics (karyotyping/FISH) and molecular testing help risk-stratify MDS (IPSS-R) and can refine prognosis and therapy choices. Flow cytometry detects PNH clones by assessing CD55/CD59 on blood cells.
- In suspected inherited marrow failure, genetic testing panels and telomere length assays are key. A careful physical exam for subtle features (nails, skin, stature, skeletal anomalies) and family history often guides which genes to test. For aplastic anaemia, severity definitions (severe vs very severe) inform urgency and whether to prioritise HSCT or immunosuppression. For MDS, IPSS-R risk groups predict survival and AML transformation risk, helping your team decide between supportive care, disease-modifying therapy, or transplant.
Treatment Options and How Doctors Decide
- Supportive care includes transfusions (red cells for anaemia; platelets for bleeding risk), antibiotics/antifungals for infections, and growth factors such as G-CSF for neutropenia in selected cases. Iron chelation (e.g., deferasirox) helps manage iron overload from chronic transfusions to protect the heart and liver. Vaccination updates, prompt fever management, and antimicrobial prophylaxis are central when neutrophils are very low.
- Immunosuppressive therapy (IST) is first-line for many with severe aplastic anaemia who lack a matched sibling donor. Standard IST uses horse anti-thymocyte globulin (ATG) plus cyclosporine; adding eltrombopag has improved response rates and durability in several studies. Response often takes months, and relapses can occur; careful monitoring and sometimes maintenance of cyclosporine are needed. Supportive care continues alongside IST.
- Hematopoietic stem cell transplantation (HSCT) offers the most definitive cure for many marrow failure syndromes, especially severe aplastic anaemia in younger patients with a matched sibling donor long-term survival frequently exceeds 80–90% in such cases. For MDS, transplant is generally reserved for higher-risk disease or selected lower-risk patients with refractory cytopenias; age, comorbidities, donor availability, and molecular risk influence timing. In inherited syndromes, reduced-intensity conditioning and tailored approaches are crucial to limit complications (e.g., in telomere disorders). Risks include graft-versus-host disease, infections, and graft failure, but outcomes continue to improve with better matching and supportive care.
- Targeted therapies for MDS include hypomethylating agents (azacitidine, decitabine) that can improve counts and delay progression, and lenalidomide, particularly effective for patients with isolated del(5q). For PNH, complement inhibitors can reduce hemolysis and thrombotic risk. A personalised path, supportive care vs IST vs HSCT vs targeted agents, depends on age, severity, donor availability, and your goals. Discuss risks, benefits, and expected timelines with your haematology team.
Living Well With Bone Marrow Failure Syndromes
- Infection prevention matters: practice good hand hygiene, avoid sick contacts during low counts, consider wearing masks in crowded indoor spaces, and keep dental care up to date to reduce infection sources. Vaccinations are important, but timing depends on treatment—live vaccines are generally avoided during significant immunosuppression or after transplant. Your team will guide which vaccines (e.g., influenza, pneumococcal) are appropriate and when. If you develop a fever during neutropenia, seek care promptly; hours matter.
- Nutrition supports energy and recovery: aim for balanced protein, fruits/vegetables (washed well), whole grains, and adequate hydration. In profound neutropenia, some centres recommend food safety precautions (well-cooked meats and eggs, careful produce washing). If you’re transfusion-dependent, discuss iron management and foods that support gut health. Moderate, tailored exercise can improve fatigue and mood; a physical therapist can help adapt plans when counts are low. Bone health may need attention if you use steroids (e.g., in DBA) or have a vitamin D deficiency. Apollo 24|7 offers home collection for vitamin D or HbA1c if you are tracking bone or metabolic health alongside treatment.
- Fertility and pregnancy planning deserve early discussion: some treatments affect fertility, and pregnancy can be a special context for marrow failure. For inherited syndromes, genetic counselling can inform family planning. Emotional well-being is integral—consider counselling, peer groups, and caregiver support. For children, coordinate school plans, infection precautions, and activity modifications so they can thrive while staying safe.
Outlook, Follow-up, and Research Frontiers
- Long-term outcomes vary by diagnosis, severity, age, and treatment choice. Severe aplastic anaemia treated with a matched-sibling HSCT often achieves excellent survival; IST responders may enjoy many years of good quality life but need monitoring for relapse and clonal evolution (development of PNH clones or progression to MDS/AML). In MDS, IPSS-R risk groups guide prognosis; lower-risk disease can be stable for years, while higher-risk disease has a greater chance of AML transformation without disease-modifying therapy or transplant.
- Follow-up typically includes periodic CBCs, iron studies (if transfused), liver/kidney function, and surveillance for clonal evolution or therapy complications. After transplant, structured monitoring looks for graft-versus-host disease, infections, and organ health. For inherited marrow failure syndromes, lifelong cancer surveillance and multisystem follow-up are essential.
- Research frontiers are encouraging: improved donor matching and conditioning regimens, novel immune-modulating therapies, complement inhibitors and beyond for PNH, targeted drugs for specific MDS subtypes, and emerging gene therapy approaches for certain inherited disorders. Telomere biology discoveries are refining treatment choices in dyskeratosis congenita and related conditions. Ask your team about clinical trials—you may access new options while advancing science.
When to See a Doctor and What to Ask?
If you develop persistent fatigue, frequent infections, unusual bruising/bleeding, or dark urine, see a clinician. A fever above 38°C (100.4°F) with low white counts is an emergency. If symptoms persist beyond two weeks, consult a doctor online with Apollo24|7 for further evaluation. They can order initial labs (CBC, iron, B12/folate) with home collection and refer you to haematology if needed.
Conclusion
Bone marrow failure syndrome is not a single disease but a family of conditions sharing one critical problem: the marrow cannot keep up with the body’s demand for healthy blood cells. The practical implications—fatigue from anaemia, infections from neutropenia, and bleeding from low platelets—can be life-altering, but they are also manageable when the cause is identified and addressed early. Acquired forms such as aplastic anaemia and myelodysplastic syndromes call for different strategies than inherited syndromes, making diagnosis and risk stratification essential first steps.
From supportive care to immunosuppressive therapy and stem cell transplantation, today’s treatments offer real hope. Many patients regain independence and good quality of life, and survival continues to improve with better donor matching, targeted therapies, and refined transplant approaches.
If symptoms persist beyond two weeks, consult a doctor online with Apollo 24|7 for further evaluation. If your condition does not improve after trying initial measures, book a physical visit to a doctor with Apollo 24|7.
Consult a Top General Practitioner for Personalised Advice
Consult a Top General Practitioner for Personalised Advice

Dr. Pinaki Mukhopadhyay
General Practitioner
33 Years • MBBS
Kolkata
MCR SUPER SPECIALITY POLY CLINIC & PATHOLOGY, Kolkata
(25+ Patients)

Dr. Sougata Kumar
General Practitioner
9 Years • MBBS
East Midnapore
VIVEKANANDA SEBA SADAN, East Midnapore

Dr Venkata Naga Sai Tribhushan Rambhatla
General Physician
3 Years • MBBS
Bengaluru
PRESTIGE SHANTHINIKETAN - SOCIETY CLINIC, Bengaluru

Dr. Arthi S
Family Physician
3 Years • MBBS
Bengaluru
PRESTIGE SHANTHINIKETAN - SOCIETY CLINIC, Bengaluru

Dr Syed Mateen Pasha
General Physician
2 Years • MBBS
Bengaluru
PRESTIGE SHANTHINIKETAN - SOCIETY CLINIC, Bengaluru
Consult a Top General Practitioner for Personalised Advice
Dr. Pinaki Mukhopadhyay
General Practitioner
33 Years • MBBS
Kolkata
MCR SUPER SPECIALITY POLY CLINIC & PATHOLOGY, Kolkata
(25+ Patients)
Dr. Sougata Kumar
General Practitioner
9 Years • MBBS
East Midnapore
VIVEKANANDA SEBA SADAN, East Midnapore
Dr Venkata Naga Sai Tribhushan Rambhatla
General Physician
3 Years • MBBS
Bengaluru
PRESTIGE SHANTHINIKETAN - SOCIETY CLINIC, Bengaluru
Dr. Arthi S
Family Physician
3 Years • MBBS
Bengaluru
PRESTIGE SHANTHINIKETAN - SOCIETY CLINIC, Bengaluru
Dr Syed Mateen Pasha
General Physician
2 Years • MBBS
Bengaluru
PRESTIGE SHANTHINIKETAN - SOCIETY CLINIC, Bengaluru
More articles from Blood Disorders

Frequently Asked Questions
Is bone marrow failure syndrome the same as leukaemia?
No. Leukaemia is a cancer of white blood cells. Bone marrow failure syndrome refers to insufficient blood cell production, which can be due to immune injury (aplastic anaemia), clonal disorders (MDS), or inherited causes. Some marrow failure states can progress to leukaemia, so monitoring is important.
How is aplastic anaemia different from MDS?
Aplastic anaemia usually shows an “empty” marrow and responds to immunosuppression or transplant. MDS shows dysplastic, often cellular marrow with genetic abnormalities and carries a risk of transforming to AML; treatment depends on IPSS-R risk and can include hypomethylating agents or transplant.
Can lifestyle changes cure bone marrow failure?
Lifestyle can’t cure marrow failure, but infection prevention, good nutrition, and safe activity can reduce complications and improve quality of life. Medical treatments—immunosuppressive therapy or stem cell transplant—are often required for severe cases.
When should I consider a bone marrow transplant?
Transplant is considered for severe aplastic anaemia (especially in younger patients with a matched sibling donor), higher-risk MDS, and many inherited syndromes. Timing depends on age, severity, donor availability, and overall health—discuss with your haematologist.
What tests diagnose inherited bone marrow failure syndromes?
Genetic testing panels, sometimes telomere length assays, and a careful exam for physical features or organ involvement help diagnose inherited syndromes like Fanconi anaemia or dyskeratosis congenita. Family counselling is often recommended.