

What Leads To Signs Of Sickle Cell Disease And Types
Learn what causes the signs of sickle cell disease, the different types like HbSS and HbSC, common triggers, complications, diagnosis, and treatment options including gene therapy.

Introduction
Sickle cell disease affects how your red blood cells carry oxygen, leading to episodes of pain, anaemia, and other complications that can impact daily life. But what exactly leads to these signs and symptoms—and how do the different types of sickle cell disease change the picture? In this guide, we break down the “why” behind the condition in clear terms, from the genetic change that causes sickling to the common triggers that set off a crisis. We also explain the main types, how doctors diagnose and monitor the condition, the latest treatments including gene therapy, and practical ways to prevent complications. Whether you are newly diagnosed, supporting a loved one, or simply want to understand this disease better, you will find step-by-step explanations and trusted guidance. If at any point symptoms are severe or unclear, seek medical help—early care makes a difference. Let’s start by understanding what sickle cell disease is and why these signs appear in the first place.
What Is Sickle Cell Disease and Why Do Signs Appear?
Here's an overview of why symptoms occur:
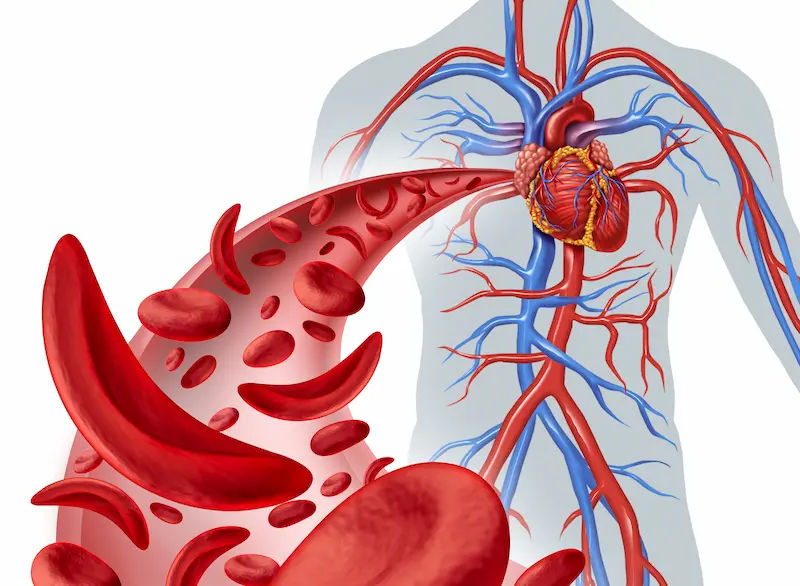
Sickle cell disease (SCD) is an inherited blood disorder caused by a change in the HBB gene that produces haemoglobin S (HbS), a variant of the oxygen-carrying protein inside red blood cells. Haemoglobin S tends to stick together under low-oxygen or stressful conditions, causing red blood cells to stiffen and distort into a “sickle” or crescent shape. Unlike healthy, flexible red blood cells that live about 120 days, sickled cells are fragile and break down early, surviving only 10–20 days. Two downstream effects explain most signs of SCD: vaso-occlusion and hemolysis.
Vaso-occlusion occurs when sickled cells clump and block small blood vessels, cutting off oxygen to tissues. This causes sudden, severe pain, acute chest syndrome when it happens in the lungs, or stroke when it affects brain blood vessels. Repeated blockages drive chronic organ stress over time.
Hemolysis happens because fragile sickled cells burst sooner. The bone marrow races to replace them but cannot keep up, leading to chronic anaemia, fatigue, pale skin, and shortness of breath. Haemoglobin breakdown raises bilirubin, causing jaundice and gallstones.
Common triggers that worsen sickling include dehydration, infection, cold temperatures, high altitude, and severe physical or emotional stress. Avoiding these triggers, staying hydrated, and treating infections promptly can reduce crises.
Consult Top Specialists
The Haemoglobin S Mutation and Inheritance
SCD follows an autosomal recessive pattern. A child inherits one HbS gene from each parent (or HbS plus another abnormal haemoglobin gene). People with sickle cell trait (one HbS and one normal gene) usually do not have the disease but can pass HbS to their children.
How Sickled Cells Cause Pain, Anaemia, and Organ Problems
Pain arises from microvascular blockages and tissue oxygen deprivation. Chronic hemolysis contributes to anaemia, jaundice, leg ulcers, and pulmonary hypertension over time.
Triggers That Worsen Sickling
Planning for hydration, vaccinations, and temperature exposure helps prevent complications. Travel and sports are possible with preparation and medical guidance.
Types of Sickle Cell Disease (and Why Type Matters)
Here's an overview of the main types:
SCD is an umbrella term for conditions where HbS is paired with another abnormal haemoglobin gene. Type can influence the frequency of pain crises, anaemia severity, and complication risk.
HbSS (“Sickle Cell Anaemia”)
- Two HbS genes. Often the most severe form.
- More frequent pain crises, higher risk of acute chest syndrome, stroke, and splenic complications in childhood.
- Typically lower baseline haemoglobin due to hemolysis.
HbSC Disease
- One HbS and one HbC gene.
- Often milder anaemia than HbSS but still at risk of pain, eye complications, avascular necrosis, and pregnancy-related issues.
- Viscosity-related complications may be more prominent than in HbSS.
Sickle Beta Thalassemia (Sβ0 and Sβ+)
- HbS with beta thalassemia mutation.
- Sβ0 thalassemia: no normal beta chain; severity often similar to HbSS.
- Sβ+ thalassemia: reduced normal beta chain; may be milder, but complications still occur.
Less Common Variants and Sickle Cell Trait
- HbSD, HbSO, HbSE can produce disease with variable severity.
- Sickle cell trait (HbAS) usually has no symptoms but can rarely cause problems under extreme conditions.
Who Is at Risk? Prevalence and Population Patterns
Here's a look at risk and distribution:
SCD is one of the most common inherited disorders worldwide, with the highest burden in sub-Saharan Africa, parts of India, the Middle East, and the Caribbean. In some regions of West and Central Africa, the sickle trait frequency can exceed 20–25%.
In the United States, about 100,000 people live with SCD, affecting approximately 1 in 365 Black or African American births and 1 in 16,300 Hispanic American births. Around 1 in 13 Black or African Americans carries the trait. The U.K. and other countries have newborn screening programs to detect SCD shortly after birth, enabling early interventions.
Global Burden
High prevalence and limited access to comprehensive care drive preventable complications. WHO emphasises newborn screening, antibiotics, vaccines, malaria prevention, and education as key interventions.
U.S./U.K. Statistics and Screening
Universal newborn screening detects SCD in most U.S. states and the U.K., allowing early treatment and parent education, which lowers early childhood deaths. Community awareness and access to specialised care remain essential.
Carriers vs Disease
Sickle cell trait is common and usually asymptomatic. Genetic counselling helps families understand inheritance risks and make informed reproductive decisions.
Signs, Symptoms, and Complications to Watch For
Here's what to look for:
SCD can affect nearly every organ system because red blood cells travel throughout the body. Symptoms often reflect both acute vaso-occlusive events and chronic hemolysis.
Common Signs
- Pain crises are hallmark events and can be triggered by dehydration, cold, infection, or sometimes no obvious cause.
- Anaemia leads to fatigue, pale skin, shortness of breath, and rapid heartbeat.
- Jaundice and dark urine result from accelerated hemolysis.
- Dactylitis (hand-foot swelling) is an early sign in infants and toddlers.
Serious Complications
- Acute chest syndrome: chest pain, cough, fever, breathing difficulty; requires urgent care.
- Stroke: particularly in children with HbSS; screening with transcranial Doppler reduces risk.
- Splenic sequestration: sudden spleen enlargement with severe anaemia; urgent care needed.
- Infections: damaged spleen increases risk of severe infections; vaccines and penicillin reduce risk.
Long-Term Organ Impacts
- Kidneys: hematuria, impaired concentration, chronic kidney disease risk.
- Eyes: proliferative retinopathy may affect vision, especially in HbSC.
- Bones/joints: avascular necrosis, chronic pain, osteoporosis.
- Pregnancy: increased risk of preeclampsia, crises, infections, fetal growth restriction, and preterm birth; specialised prenatal care improves outcomes.
Seek emergency care for severe chest pain, breathing difficulty, stroke symptoms, high fever, or rapidly enlarging abdomen in a child. For recurring or persistent symptoms, consult a doctor online with Apollo 24|7.
Diagnosis: How SCD Is Found and Monitored
Here's how SCD is diagnosed and tracked:
Most people in countries with newborn screening are diagnosed shortly after birth. Elsewhere, diagnosis may happen after a pain crisis or severe anaemia. Early diagnosis enables interventions that prevent complications.
Newborn Screening and Confirmatory Tests
- Newborn screening detects abnormal haemoglobins using isoelectric focusing or HPLC.
- Confirmatory testing includes haemoglobin electrophoresis or HPLC; DNA testing may clarify complex cases.
Labs and Imaging
- CBC shows anaemia; reticulocyte count is elevated.
- Bilirubin and LDH rise due to hemolysis.
- Transcranial Doppler (TCD) screens children for stroke risk.
- Additional assessments include echocardiography, eye exams, kidney function tests, and MRI when needed.
When to Seek Urgent Care
- Fever ≥38.5°C, chest pain/shortness of breath, stroke symptoms, severe abdominal swelling, or uncontrolled pain.
- Routine care: regular haematology visits for medications, growth tracking, vaccines, and screening.
Apollo 24|7 offers home collection for CBC and bilirubin; confirmatory tests may need specialised lab referral.
Treatment Options Today: From Medicines to Curative Therapies
Here's how treatment is approached:
Disease-Modifying Treatments
- Hydroxyurea: increases fetal haemoglobin to reduce sickling and crises.
- L-glutamine: reduces oxidative stress in red cells.
- Voxelotor: improves haemoglobin’s oxygen affinity to reduce polymerisation.
- Crizanlizumab: reduces cell adhesion to prevent vaso-occlusive crises.
Transfusions, Iron Chelation, and Crisis Management
- Transfusions lower HbS percentage for acute or preventive use.
- Iron overload from transfusions requires chelation.
- Crisis management includes hydration, pain control, oxygen if needed, and infection treatment.
Curative Options
- A hematopoietic stem cell transplant can cure SCD in selected patients.
- Gene therapy for SCD has been approved for certain patients to induce fetal haemoglobin or correct the defect.
Daily Management and Prevention: Living Well with SCD
Here's how to manage day-to-day:
Vaccines, Penicillin, Hydration, Trigger Avoidance
- Keep vaccinations up to date and take penicillin in early childhood.
- Maintain hydration, plan for temperature changes, and discuss high-altitude travel with a clinician.
Pain Management Plan
- Home: NSAIDs, prescribed medications, heat packs, and rest.
- Hospital: seek care if pain is uncontrolled, fever develops, or chest symptoms appear.
- Keep a crisis kit with medications, water, comfort items, and a written pain plan.
Nutrition, Exercise, Mental Health, and Social Support
- Balanced diet with folate-rich foods; monitor vitamin D and bone health.
- Gentle exercise with rest and hydration.
- Counselling, peer support, and workplace/school accommodations improve quality of life.
Special Situations Across Life Stages
Here's what to consider:
Children and Teens
- Monitor growth and nutrition.
- Individual care plans for school, hydration breaks, and PE participation.
- Gradual transition to adult care improves self-advocacy and medication management.
Pregnancy and Family Planning
- Preconception counselling for risks, vaccines, folic acid, and medications.
- Close obstetric-hematology care during pregnancy; consider low-dose aspirin or transfusions if advised.
Surgery, Anaesthesia, Travel, and High-Altitude Considerations
- Pre-op assessment and possible transfusion reduce perioperative risks.
- Travel plans should include hydration, rest, and oxygen if needed; avoid extreme cold.
Myths vs Facts About Sickle Cell Disease
- SCD affects multiple populations worldwide, not only people of African descent.
- Pain crises can occur without obvious triggers.
- Trait and disease are not the same; trait usually has no symptoms.
- Vaccines, penicillin, hydroxyurea, transfusions, and gene therapy can prevent complications.
Questions to Ask Your Doctor
- What type of sickle cell disease do I have, and how does it affect my risks?
- Should I start or adjust disease-modifying therapy?
- What is my crisis plan at home and when to go to hospital?
- Do I need screening tests this year?
- Am I a candidate for transfusions, stem cell transplant, or gene therapy?
- How should I plan for pregnancy, travel, or surgery?
- What vaccines or antibiotics do I need now?
Conclusion
Sickle cell disease is complex, but its signs become clearer when you understand the chain of events: a genetic change causes red cells to sickle, which leads to vessel blockages, pain, organ stress, and ongoing anaemia. The type—HbSS, HbSC, Sβ0, Sβ+, or other variants—affects risk, yet personal factors and access to care are equally important. Today’s tools—from newborn screening and vaccines to hydroxyurea, newer therapies, and curative options—can reduce crises and improve quality of life. Daily habits such as staying hydrated, keeping warm, and acting early when unwell make a real difference, alongside a clear crisis plan and regular check-ins with your care team. Apollo 24|7 offers online consultations and home collection for tests like CBC and bilirubin. With knowledge, preparation, and strong healthcare support, living well with sickle cell disease is achievable. Use the checklist above, bring questions to appointments, and connect with support networks—you are not alone.
Consult Top Specialists
Consult Top Specialists

Dr Vijeya Lakshmi Chakravarthy
General Physician
3 Years • MBBS
Bengaluru
PRESTIGE SHANTHINIKETAN - SOCIETY CLINIC, Bengaluru

Dr. Thorana Prakash M
General Physician
2 Years • MBBS
Bengaluru
PRESTIGE SHANTHINIKETAN - SOCIETY CLINIC, Bengaluru

Dr. Ramalinga Reddy
General Physician
5 Years • MBBS MD General medicine
Bengaluru
PRESTIGE SHANTHINIKETAN - SOCIETY CLINIC, Bengaluru

Dr Aniruddha Burli
Haematologist
5 Years • MBBS, MD (General Medicine) ,DM (Clinical Hematology, AIIMS New Delhi)
Bengaluru
Apollo Hospitals Bannerghatta Road, Bengaluru

Dr. E Prabhakar Sastry
General Physician/ Internal Medicine Specialist
40 Years • MD(Internal Medicine)
Manikonda Jagir
Apollo Clinic, Manikonda, Manikonda Jagir
(200+ Patients)
Consult Top Specialists
Dr Vijeya Lakshmi Chakravarthy
General Physician
3 Years • MBBS
Bengaluru
PRESTIGE SHANTHINIKETAN - SOCIETY CLINIC, Bengaluru
Dr. Thorana Prakash M
General Physician
2 Years • MBBS
Bengaluru
PRESTIGE SHANTHINIKETAN - SOCIETY CLINIC, Bengaluru
Dr. Ramalinga Reddy
General Physician
5 Years • MBBS MD General medicine
Bengaluru
PRESTIGE SHANTHINIKETAN - SOCIETY CLINIC, Bengaluru
Dr Aniruddha Burli
Haematologist
5 Years • MBBS, MD (General Medicine) ,DM (Clinical Hematology, AIIMS New Delhi)
Bengaluru
Apollo Hospitals Bannerghatta Road, Bengaluru
Dr. E Prabhakar Sastry
General Physician/ Internal Medicine Specialist
40 Years • MD(Internal Medicine)
Manikonda Jagir
Apollo Clinic, Manikonda, Manikonda Jagir
(200+ Patients)
More articles from Sickle Cell Anemia disease
Frequently Asked Questions
Is sickle cell trait the same as sickle cell disease?
No. Sickle cell trait usually causes no symptoms, while sickle cell disease can cause pain crises, anaemia, and complications.
What triggers a sickle cell crisis and how can I prevent it?
Dehydration, cold, infection, stress, and high altitude are common triggers. Hydrate, stay warm, keep up to date on vaccines, and plan travel.
How is sickle cell disease diagnosed?
Newborn screening followed by haemoglobin electrophoresis or HPLC confirms the diagnosis and type. CBC, reticulocyte counts, bilirubin, and transcranial Doppler screening are used in children.
Are there cures for sickle cell disease?
Stem cell transplant can cure some patients. Gene therapy has also been approved for certain patients. Discuss benefits and risks with a specialist.
What is acute chest syndrome?
A serious complication with chest pain, cough, fever, and breathing difficulty plus lung changes on imaging. Urgent hospital care is required; book a visit with Apollo 24|7 if needed.